Exploring Alphafold Model (Part 2)
I've written previously about my enthusiasm for implementing and using Deepmind's Alphafold model for protein folding. AlphaFold2 finally achieved what many thought impossible: predicting protein structures with near-experimental accuracy from sequence alone. But as an independent researcher without access to massive computational resources, I needed a more accessible way to harness this technology. Fortunately I have discovered ColabFold, a brilliant adaptation that makes cutting-edge protein structure prediction easy for just about anyone with an internet connection.
ColabFold
My first experience with ColabFold was one of the easiest experiences I've ever had with coding. I navigated to the notebook, pasted in the protein sequence I'd worked with previously, clicked "Run all," and within minutes, I was looking at a detailed 3D model that would have taken years to determine experimentally. While the interface is deceptively simple, under the hood incredibly sophisticated analysis is happening.
Preparing My Experiment
For my first prediction, I decided to use a relatively small protein with 56 amino acids:
PIAQIHILEGRSDEQKETLIREVSEAISRSLDAPLTSVRVIITEMAKGHFGIGGELASK
This is a bacterial transcription factor that I've been studying for a while, but whose structure remains experimentally undetermined. One of the aspects I appreciate about ColabFold is how it handles various options. I decided not to use AMBER relaxation or any templates initially—meaning the prediction would be based purely on the sequence and evolutionary information, without reference to any known structures.
Behind the scenes, ColabFold installs all the necessary dependencies—and there are quite a few. Considering i spent the better part of a week implementing all the dependencies when I first started with Alphafold, shielding users from that complexity in this way is a pretty great feature of ColabFold.
Generating Multiple Sequence Alignments
This is where ColabFold truly shines compared to the original AlphaFold implementation. The standard AlphaFold pipeline uses HHblits to search against enormous databases like BFD and MGnify, which can take hours or days and requires terabytes of storage.
ColabFold uses MMseqs2, a much faster homology detection method, searching against databases like UniRef and environmental sequences. For my protein, I chose "mmseqs2_uniref_env" which searches both UniRef and environmental sequence databases for homologs. Again, a million times faster than my first implementation.
The evolutionary information captured in these Multiple Sequence Alignments (MSAs) is crucial for accurate prediction. As proteins evolve, certain positions change in coordinated ways to maintain structure and function. These coevolutionary patterns provide powerful clues about which amino acids are likely to be spatially close in the folded protein. When I ran this step, ColabFold found hundreds of related sequences for my protein—a good sign that the prediction would be reliable. I could see this visually in the MSA coverage plot that showed evolutionary conservation across the protein's length.

Running the Prediction
Time for the meat of the analysis. ColabFold feeds the MSA and configuration into AlphaFold's neural networks, which have been trained on the entire Protein Data Bank plus additional structures. For my protein, the process took only about 5 minutes on the Google Colab GPU—orders of magnitude faster than experimental methods and even multiple times faster than my previous Alphafold implementation.
When the prediction finished, I was greeted with a stunning 3D visualization of my protein, colored by confidence. The predicted structure showed a compact globular fold with several alpha helices—typical for a DNA-binding protein.
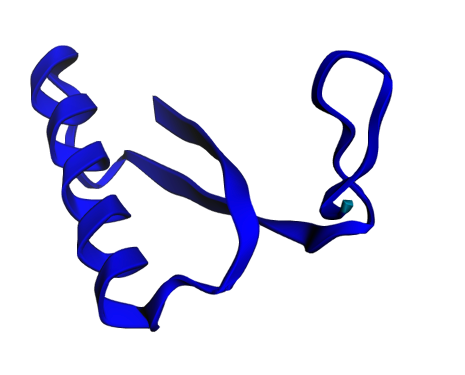
In addition pretty visualization; it provides rich data to assess the reliability of the prediction. The model produces a per-residue confidence score called pLDDT (predicted Local Distance Difference Test), ranging from 0-100, with higher values indicating greater confidence. My protein showed high confidence (70-90) across most of its length, with slightly lower confidence at the termini—exactly what you'd expect, since protein ends usually have more iterations.
The visualization uses color coding to make this immediately apparent:
Blue regions (90-100): Very high confidence
Light blue (70-90): Confident
Yellow (50-70): Low confidence
Red (<50): Very low confidence
For multimeric proteins, ColabFold also provides Predicted Aligned Error (PAE) plots that show confidence in the relative positioning of residues—crucial for assessing interface quality in protein complexes.
ColabFold something that surprised me is that it didn't just give me one prediction; it provided five ranked models. I could examine each one by changing the rank number in the visualization cell. For my protein, the top-ranked model had the highest average pLDDT score, but the other models showed similar overall folds with minor variations in loop regions—consistent with what we know about protein dynamics.
Final Thoughts
Exciting as all of this is, I had to ask myself if this was categorically better than Alphafold? And the more i learned the more complicated that answer got. One limitation is that Google Colab assigns different GPUs with varying memory limits, so sometimes a long protein or complex will exceed available memory. Also while MMseqs2 is faster, it sometimes finds fewer homologous sequences than the full AlphaFold pipeline which includes other databases, which could affect the prediction accuracy.
It seems like ColabFold sacrifices a little bit of accuracy for a lot of speed, ease, and meta-analysis. Which is great! But if I'm entering into a competition or writing a research paper, I'd probably still use the full AlphaFold pipeline locally to get the best results.
But if lime me you're exploring protein folding as a passion project, or even if you're actually working with proteins in any capacity—whether you're a researcher, student, or educator—I highly recommend giving ColabFold a try. The barrier to entry is minimal, and the potential insights are enormous. As the authors of ColabFold eloquently put it, their goal is "making protein folding accessible to all." In my experience, they've succeeded brilliantly.
As always my code can be found on my GitHub here.








