Dimensionality Expansion for Genome Sequencing
Joining my dimensionality expansion methods to my bioinformatics research
As data scientists, we've all hit that frustrating wall when working with language models or sequence analysis—the dreaded sequence length limitation. I've spent countless hours optimizing attention mechanisms and tweaking model architectures, only to gain marginal improvements in handling longer texts or genomic sequences. But what if we've been tackling this problem from the wrong angle all along?
Breaking Free from One-Dimensional Thinking
Recently, I've been exploring an alternative approach that has transformed how I work with large sequences. Rather than continually optimizing model architectures (which seems to be where most research focuses), I started questioning our fundamental representation of sequential data.
Think about it: why do we insist on representing sequences as one-dimensional arrays? In the physical world, we don't store large amounts of text in a single, unbroken line. Instead, we arrange words on pages and pages in books. This dimensional organization isn't just convenient—it's efficient.
This realization led me to a simple but powerful insight: what if we increased the dimensionality of our sequence representation?
From Lines to Planes to Volumes
Let me walk you through the conceptual progression: A single sentence is essentially a one-dimensional representation—a linear sequence of tokens. A page of text is two-dimensional, organizing that sequence into rows and columns. A book takes this further into three dimensions, stacking those pages into a compact volume.
With each increase in dimensionality, we gain a more efficient way to represent and transport the same information. This isn't just a physical convenience—it suggests a fundamentally different way to encode data for our models.
The Perfect Testing Ground: Biological Sequences
While working on a genomics project, I realized biological sequences offer the perfect testing ground for this approach for two key reasons. First, we have access to vast amounts of genomic data; and second, biological sequences (particularly DNA and RNA) have a remarkably limited alphabet. The four-letter alphabet of DNA (A, T, G, C) is particularly appealing from an encoding perspective. We can easily one-hot encode each nucleotide, but the magic happens in how we arrange these encodings. Instead of feeding a genomic sequence to a model as a 1D array, I reshape it into a 2D array—essentially creating an "image" where the (x,y) position encodes the sequence order and the channel dimension encodes the specific nucleotide at that position
Putting Theory Into Practice: The HIV Genome
To test this approach, I downloaded HIV genome sequences from the NCBI database. The average HIV genome is around 9,000 bases long, which conveniently reshapes into a 24×24×16 array (plus our channel dimension for the nucleotide encoding).
Since the final array has more elements than the typical genome, I applied a small amount of padding. With this transformation complete, I could now leverage architectures designed for image processing!

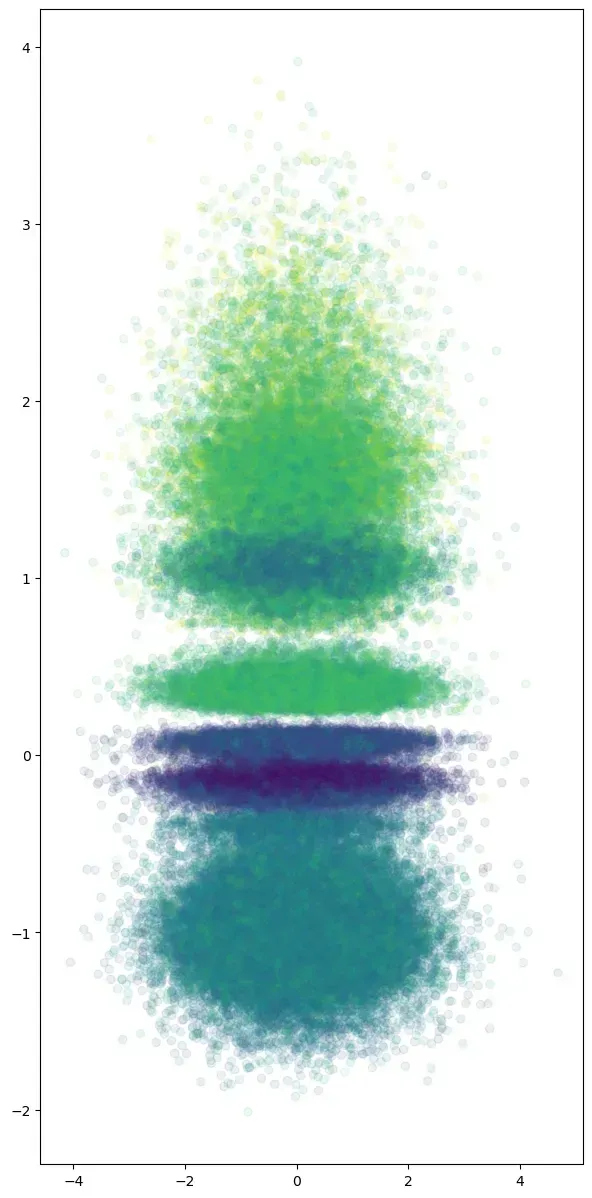
I trained a variational convolutional autoencoder on this reshaped data, which not only performed dimensionality reduction but also naturally clustered similar sequences together—all without requiring labeled data.

The most exciting part? The generative component of the model successfully reconstructed sequences, allowing us to compare them against fragments and evaluate reconstruction quality.

Expanding to Dengue and SARS-CoV-2
Encouraged by these results, I applied the same approach to Dengue virus genomes and observed similar clustering by sequence similarity.

The flexibility offered by expanding the number of dimensions results in a quick and simple method to analyze large sequences even when the dataset does not fit into memory. Data transformation can be done relatively quickly and the genomic sequences can be fed to the network by loading them in batches. This specific case can be found by analyzing the current pandemic virus.

The real test came when working with SARS-CoV-2 data. Given the volume of sequences, I implemented a batch processing approach where genomes were loaded, transformed, and fed to a convolutional autoencoder on the fly.


This not only clustered the sequences effectively but revealed something fascinating—distinct mutation patterns specific to each cluster. When visualizing the changes across the dimension that sorts the sequences, these patterns became clearly visible, suggesting specific evolutionary pathways the virus follows to adapt to environmental pressures.

Why This Matters for Data Scientists
This dimensional transformation approach offers several practical advantages for anyone working with large sequential data. Perhaps most importantly, it demonstrates how rethinking data representation can sometimes be more productive than endlessly tweaking model architectures.
While I've focused on genomic applications, this approach has potential anywhere we deal with long sequences including protein structures which I'm excited to get into in the near future. The key insight is that dimensionality can be a feature, not a limitation. By reshaping our data thoughtfully, we open up new architectural possibilities and processing efficiencies.
More broadly, I believe we've only scratched the surface of what's possible with dimensional transformations of sequential data. As we continue to face challenges with increasingly large sequences, rethinking our fundamental data structures may prove more fruitful than continually scaling model architectures.